
Autophagy increase in Merosin-Deficient Congenital Muscular Dystrophy type 1A
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Published: 28 July 2023
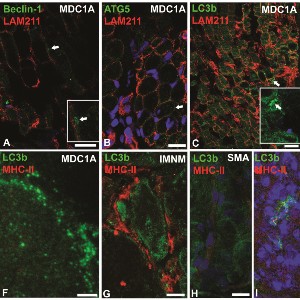
The autophagy process recycles dysfunctional cellular components and protein aggregates by sequestering them in autophagosomes directed to lysosomes for enzymatic degradation. A basal level of autophagy is essential for skeletal muscle maintenance. Increased autophagy occurs in several forms of muscular dystrophy and in the merosin-deficient congenital muscular dystrophy 1A mouse model (dy3k/dy3k) lacking the laminin-α2 chain. This pilot study aimed to compare autophagy marker expression and autophagosomes presence using light and electron microscopes and western blotting in diagnostic muscle biopsies from newborns affected by different congenital muscular myopathies and dystrophies. Morphological examination showed dystrophic muscle features, predominance of type 2A myofibers, accumulation of autophagosomes in the subsarcolemmal areas, increased number of autophagosomes overexpressing LC3b, Beclin-1 and ATG5, in the merosin-deficient newborn suggesting an increased autophagy. In Duchenne muscular dystrophy, nemaline myopathy, and spinal muscular atrophy the predominant accumulation of p62+ puncta rather suggests an autophagy impairment.
Downloads
Dubowitz V. 22nd ENMC sponsored workshop on congenital muscular dystrophy held in Baarn, The Netherlands, 14-16 May 1993. Neuromuscul Disord. 1994 Jan;4(1):75-81. PMID: 8173355. DOI: https://doi.org/10.1016/0960-8966(94)90051-5
Graziano A, Bianco F, D'Amico A, Moroni I, Messina S, Bruno C, Pegoraro E, Mora M, Astrea G, Magri F, Comi GP, Berardinelli A, Moggio M, Morandi L, Pini A, Petillo R, Tasca G, Monforte M, Minetti C, Mongini T, Ricci E, Gorni K, Battini R, Villanova M, Politano L, Gualandi F, Ferlini A, Muntoni F, Santorelli FM, Bertini E, Pane M, Mercuri E. Prevalence of congenital muscular dystrophy in Italy: a population study. Neurology. 2015 Mar 3;84(9):904-11. Epub 2015 Feb 4. PMID: 25653289; PMCID: PMC4351663. DOI: https://doi.org/10.1212/WNL.0000000000001303
Tomé FM, Evangelista T, Leclerc A, Sunada Y, Manole E, Estournet B, Barois A, Campbell KP, Fardeau M. Congenital muscular dystrophy with merosin deficiency. C R Acad Sci III. 1994 Apr;317(4):351-7. PMID: 8000914.
Zambon AA, Muntoni F. Congenital muscular dystrophies: What is new? Neuromuscul Disord. 2021 Oct;31(10):931-942. Epub 2021 Jul 28. PMID: 34470717. DOI: https://doi.org/10.1016/j.nmd.2021.07.009
Helbling-Leclerc A, Zhang X, Topaloglu H, Cruaud C, Tesson F, Weissenbach J, Tomé FM, Schwartz K, Fardeau M, Tryggvason K, et al. Mutations in the laminin alpha 2-chain gene (LAMA2) cause merosin-deficient congenital muscular dystrophy. Nat Genet. 1995 Oct;11(2):216-8. PMID: 7550355. DOI: https://doi.org/10.1038/ng1095-216
Huang K, Bi FF, Yang H. A Systematic Review and Meta-Analysis of the Prevalence of Congenital Myopathy. Front Neurol. 2021 Nov 2;12:761636. Erratum in: Front Neurol. 2022 Feb 14;13:857959. PMID: 34795634; PMCID: PMC8592924.
Carmignac V, Svensson M, Körner Z, Elowsson L, Matsumura C, Gawlik KI, Allamand V, Durbeej M. Autophagy is increased in laminin α2 chain-deficient muscle and its inhibition improves muscle morphology in a mouse model of MDC1A. Hum Mol Genet. 2011 Dec 15;20(24):4891-902. Epub 2011 Sep 14. PMID: 21920942. DOI: https://doi.org/10.1093/hmg/ddr427
Girolamo F, Lia A, Annese T, Giannini M, Amati A, D'Abbicco D, Tampoia M, Virgintino D, Ribatti D, Serlenga L, Iannone F, Trojano M. Autophagy markers LC3 and p62 accumulate in immune-mediated necrotizing myopathy. Muscle Nerve. 2019 Sep;60(3):315-327. doi: 10.1002/mus.26608. Epub 2019 Jun 21. PMID: 31172530. DOI: https://doi.org/10.1002/mus.26608
Girolamo F, Lia A, Amati A, Strippoli M, Coppola C, Virgintino D, Roncali L, Toscano A, Serlenga L, Trojano M. Overexpression of autophagic proteins in the skeletal muscle of sporadic inclusion body myositis. Neuropathol Appl Neurobiol. 2013 Dec;39(7):736-49. DOI: https://doi.org/10.1111/nan.12040
Lia A, Annese T, Fornaro M, Giannini M, D'Abbicco D, Errede M, Lorusso L, Amati A, Tampoia M, Trojano M, Virgintino D, Ribatti D, Serlenga L, Iannone F, Girolamo F. Perivascular and endomysial macrophages expressing VEGF and CXCL12 promote angiogenesis in anti-HMGCR immune-mediated necrotizing myopathy. Rheumatology (Oxford). 2022 Aug 3;61(8):3448-3460. PMID: 34864921. DOI: https://doi.org/10.1093/rheumatology/keab900
Pontrelli P, Oranger A, Barozzino M, Divella C, Conserva F, Fiore MG, Rossi R, Papale M, Castellano G, Simone S, Laviola L, Giorgino F, Piscitelli D, Gallone A, Gesualdo L. Deregulation of autophagy under hyperglycemic conditions is dependent on increased lysine 63 ubiquitination: a candidate mechanism in the progression of diabetic nephropathy. J Mol Med (Berl). 2018 Jul;96(7):645-659. Epub 2018 May 27. PMID: 29806072. DOI: https://doi.org/10.1007/s00109-018-1656-3
Desantis S, Accogli G, Burk J, Zizza S, Mastrodonato M, Francioso EG, Rossi R, Crovace A, Resta L. Ultrastructural characteristics of ovine bone marrow-derived mesenchymal stromal cells cultured with a silicon stabilized tricalcium phosphate bioceramic. Microsc Res Tech. 2017 Nov;80(11):1189-1198. Epub 2017 Aug 11. PMID: 28799674. DOI: https://doi.org/10.1002/jemt.22916
Bennington JL, Krupp M. Morphometric analysis of muscle. In: Muscle pathology, ed. Heffner, Churchill Livingstone, New York, 1984: p. 43-71.
Zhao T, Zheng T, Yu H, Hu BH, Hu B, Ma P, Yang Y, Yang N, Hu J, Cao T, Chen G, Yan B, Peshoff M, Hatzoglou M, Geng R, Li B, Zheng QY. Autophagy impairment as a key feature for acetaminophen-induced ototoxicity. Cell Death Dis. 2021 Jan 4;12(1):3. PMID: 33414397; PMCID: PMC7791066. DOI: https://doi.org/10.1038/s41419-020-03328-6
Nascimbeni AC, Fanin M, Masiero E, Angelini C, Sandri M. Impaired autophagy contributes to muscle atrophy in glycogen storage disease type II patients. Autophagy. 2012 Nov;8(11):1697-700. Epub 2012 Aug 31. PMID: 22940840; PMCID: PMC3494606. DOI: https://doi.org/10.4161/auto.21691
Fan L, Yin S, Zhang E, Hu H. Role of p62 in the regulation of cell death induction. Apoptosis. 2018 Apr;23(3-4):187-193. PMID: 29480462. DOI: https://doi.org/10.1007/s10495-018-1445-z
Klionsky DJ, Abdel-Aziz AK, Abdelfatah S, Abdellatif M, Abdoli A, Abel S, Abeliovich H, Abildgaard MH, Abudu YP, Acevedo-Arozena A, Adamopoulos IE, Adeli K, Adolph TE, Adornetto A, Aflaki E, Agam G, Agarwal A, Aggarwal BB, Agnello M, Agostinis P, Agrewala JN, Agrotis A, Aguilar PV, Ahmad ST, Ahmed ZM, Ahumada-Castro U, Aits S, Aizawa S, Akkoc Y, Akoumianaki T, Akpinar HA, Al-Abd AM, Al-Akra L, Al-Gharaibeh A, Alaoui-Jamali MA, Alberti S, Alcocer-Gómez E, Alessandri C, Ali M, Alim Al-Bari MA, Aliwaini S, Alizadeh J, Almacellas E, Almasan A, Alonso A, Alonso GD, Altan-Bonnet N, Altieri DC, Álvarez ÉMC, Alves S, Alves da Costa C, Alzaharna MM, Amadio M, Amantini C, Amaral C, Ambrosio S, Amer AO, Ammanathan V, An Z, Andersen SU, Andrabi SA, Andrade-Silva M, Andres AM, Angelini S, Ann D, Anozie UC, Ansari MY, Antas P, Antebi A, Antón Z, Anwar T, Apetoh L, Apostolova N, Araki T, Araki Y, Arasaki K, Araújo WL, Araya J, Arden C, Arévalo MA, Arguelles S, Arias E, Arikkath J, Arimoto H, Ariosa AR, Armstrong-James D, Arnauné-Pelloquin L, Aroca A, Arroyo DS, Arsov I, Artero R, Asaro DML, Aschner M, Ashrafizadeh M, Ashur-Fabian O, Atanasov AG, Au AK, Auberger P, Auner HW, Aurelian L, Autelli R, Avagliano L, Ávalos Y, Aveic S, Aveleira CA, Avin-Wittenberg T, Aydin Y, Ayton S, Ayyadevara S, Azzopardi M, Baba M, Backer JM, Backues SK, Bae DH, Bae ON, Bae SH, Baehrecke EH, Baek A, Baek SH, Baek SH, Bagetta G, Bagniewska-Zadworna A, Bai H, Bai J, Bai X, Bai Y, Bairagi N, Baksi S, Balbi T, Baldari CT, Balduini W, Ballabio A, Ballester M, Balazadeh S, Balzan R, Bandopadhyay R, Banerjee S, Banerjee S, Bánréti Á, Bao Y, Baptista MS, Baracca A, Barbati C, Bargiela A, Barilà D, Barlow PG, Barmada SJ, Barreiro E, Barreto GE, Bartek J, Bartel B, Bartolome A, Barve GR, Basagoudanavar SH, Bassham DC, Bast RC Jr, Basu A, Batoko H, Batten I, Baulieu EE, Baumgarner BL, Bayry J, Beale R, Beau I, Beaumatin F, Bechara LRG, Beck GR Jr, Beers MF, Begun J, Behrends C, Behrens GMN, Bei R, Bejarano E, Bel S, Behl C, Belaid A, Belgareh-Touzé N, Bellarosa C, Belleudi F, Belló Pérez M, Bello-Morales R, Beltran JSO, Beltran S, Benbrook DM, Bendorius M, Benitez BA, Benito-Cuesta I, Bensalem J, Berchtold MW, Berezowska S, Bergamaschi D, Bergami M, Bergmann A, Berliocchi L, Berlioz-Torrent C, Bernard A, Berthoux L, Besirli CG, Besteiro S, Betin VM, Beyaert R, Bezbradica JS, Bhaskar K, Bhatia-Kissova I, Bhattacharya R, Bhattacharya S, Bhattacharyya S, Bhuiyan MS, Bhutia SK, Bi L, Bi X, Biden TJ, Bijian K, Billes VA, Binart N, Bincoletto C, Birgisdottir AB, Bjorkoy G, Blanco G, Blas-Garcia A, Blasiak J, Blomgran R, Blomgren K, Blum JS, Boada-Romero E, Boban M, Boesze-Battaglia K, Boeuf P, Boland B, Bomont P, Bonaldo P, Bonam SR, Bonfili L, Bonifacino JS, Boone BA, Bootman MD, Bordi M, Borner C, Bornhauser BC, Borthakur G, Bosch J, Bose S, Botana LM, Botas J, Boulanger CM, Boulton ME, Bourdenx M, Bourgeois B, Bourke NM, Bousquet G, Boya P, Bozhkov PV, Bozi LHM, Bozkurt TO, Brackney DE, Brandts CH, Braun RJ, Braus GH, Bravo-Sagua R, Bravo-San Pedro JM, Brest P, Bringer MA, Briones-Herrera A, Broaddus VC, Brodersen P, Brodsky JL, Brody SL, Bronson PG, Bronstein JM, Brown CN, Brown RE, Brum PC, Brumell JH, Brunetti-Pierri N, Bruno D, Bryson-Richardson RJ, Bucci C, Buchrieser C, Bueno M, Buitrago-Molina LE, Buraschi S, Buch S, Buchan JR, Buckingham EM, Budak H, Budini M, Bultynck G, Burada F, Burgoyne JR, Burón MI, Bustos V, Büttner S, Butturini E, Byrd A, Cabas I, Cabrera-Benitez S, Cadwell K, Cai J, Cai L, Cai Q, Cairó M, Calbet JA, Caldwell GA, Caldwell KA, Call JA, Calvani R, Calvo AC, Calvo-Rubio Barrera M, Camara NO, Camonis JH, Camougrand N, Campanella M, Campbell EM, Campbell-Valois FX, Campello S, Campesi I, Campos JC, Camuzard O, Cancino J, Candido de Almeida D, Canesi L, Caniggia I, Canonico B, Cantí C, Cao B, Caraglia M, Caramés B, Carchman EH, Cardenal-Muñoz E, Cardenas C, Cardenas L, Cardoso SM, Carew JS, Carle GF, Carleton G, Carloni S, Carmona-Gutierrez D, Carneiro LA, Carnevali O, Carosi JM, Carra S, Carrier A, Carrier L, Carroll B, Carter AB, Carvalho AN, Casanova M, Casas C, Casas J, Cassioli C, Castillo EF, Castillo K, Castillo-Lluva S, Castoldi F, Castori M, Castro AF, Castro-Caldas M, Castro-Hernandez J, Castro-Obregon S, Catz SD, Cavadas C, Cavaliere F, Cavallini G, Cavinato M, Cayuela ML, Cebollada Rica P, Cecarini V, Cecconi F, Cechowska-Pasko M, Cenci S, Ceperuelo-Mallafré V, Cerqueira JJ, Cerutti JM, Cervia D, Cetintas VB, Cetrullo S, Chae HJ, Chagin AS, Chai CY, Chakrabarti G, Chakrabarti O, Chakraborty T, Chakraborty T, Chami M, Chamilos G, Chan DW, Chan EYW, Chan ED, Chan HYE, Chan HH, Chan H, Chan MTV, Chan YS, Chandra PK, Chang CP, Chang C, Chang HC, Chang K, Chao J, Chapman T, Charlet-Berguerand N, Chatterjee S, Chaube SK, Chaudhary A, Chauhan S, Chaum E, Checler F, Cheetham ME, Chen CS, Chen GC, Chen JF, Chen LL, Chen L, Chen L, Chen M, Chen MK, Chen N, Chen Q, Chen RH, Chen S, Chen W, Chen W, Chen XM, Chen XW, Chen X, Chen Y, Chen YG, Chen Y, Chen Y, Chen YJ, Chen YQ, Chen ZS, Chen Z, Chen ZH, Chen ZJ, Chen Z, Cheng H, Cheng J, Cheng SY, Cheng W, Cheng X, Cheng XT, Cheng Y, Cheng Z, Chen Z, Cheong H, Cheong JK, Chernyak BV, Cherry S, Cheung CFR, Cheung CHA, Cheung KH, Chevet E, Chi RJ, Chiang AKS, Chiaradonna F, Chiarelli R, Chiariello M, Chica N, Chiocca S, Chiong M, Chiou SH, Chiramel AI, Chiurchiù V, Cho DH, Choe SK, Choi AMK, Choi ME, Choudhury KR, Chow NS, Chu CT, Chua JP, Chua JJE, Chung H, Chung KP, Chung S, Chung SH, Chung YL, Cianfanelli V, Ciechomska IA, Cifuentes M, Cinque L, Cirak S, Cirone M, Clague MJ, Clarke R, Clementi E, Coccia EM, Codogno P, Cohen E, Cohen MM, Colasanti T, Colasuonno F, Colbert RA, Colell A, Čolić M, Coll NS, Collins MO, Colombo MI, Colón-Ramos DA, Combaret L, Comincini S, Cominetti MR, Consiglio A, Conte A, Conti F, Contu VR, Cookson MR, Coombs KM, Coppens I, Corasaniti MT, Corkery DP, Cordes N, Cortese K, Costa MDC, Costantino S, Costelli P, Coto-Montes A, Crack PJ, Crespo JL, Criollo A, Crippa V, Cristofani R, Csizmadia T, Cuadrado A, Cui B, Cui J, Cui Y, Cui Y, Culetto E, Cumino AC, Cybulsky AV, Czaja MJ, Czuczwar SJ, D'Adamo S, D'Amelio M, D'Arcangelo D, D'Lugos AC, D'Orazi G, da Silva JA, Dafsari HS, Dagda RK, Dagdas Y, Daglia M, Dai X, Dai Y, Dai Y, Dal Col J, Dalhaimer P, Dalla Valle L, Dallenga T, Dalmasso G, Damme M, Dando I, Dantuma NP, Darling AL, Das H, Dasarathy S, Dasari SK, Dash S, Daumke O, Dauphinee AN, Davies JS, Dávila VA, Davis RJ, Davis T, Dayalan Naidu S, De Amicis F, De Bosscher K, De Felice F, De Franceschi L, De Leonibus C, de Mattos Barbosa MG, De Meyer GRY, De Milito A, De Nunzio C, De Palma C, De Santi M, De Virgilio C, De Zio D, Debnath J, DeBosch BJ, Decuypere JP, Deehan MA, Deflorian G, DeGregori J, Dehay B, Del Rio G, Delaney JR, Delbridge LMD, Delorme-Axford E, Delpino MV, Demarchi F, Dembitz V, Demers ND, Deng H, Deng Z, Dengjel J, Dent P, Denton D, DePamphilis ML, Der CJ, Deretic V, Descoteaux A, Devis L, Devkota S, Devuyst O, Dewson G, Dharmasivam M, Dhiman R, di Bernardo D, Di Cristina M, Di Domenico F, Di Fazio P, Di Fonzo A, Di Guardo G, Di Guglielmo GM, Di Leo L, Di Malta C, Di Nardo A, Di Rienzo M, Di Sano F, Diallinas G, Diao J, Diaz-Araya G, Díaz-Laviada I, Dickinson JM, Diederich M, Dieudé M, Dikic I, Ding S, Ding WX, Dini L, Dinić J, Dinic M, Dinkova-Kostova AT, Dionne MS, Distler JHW, Diwan A, Dixon IMC, Djavaheri-Mergny M, Dobrinski I, Dobrovinskaya O, Dobrowolski R, Dobson RCJ, Đokić J, Dokmeci Emre S, Donadelli M, Dong B, Dong X, Dong Z, Dorn Ii GW, Dotsch V, Dou H, Dou J, Dowaidar M, Dridi S, Drucker L, Du A, Du C, Du G, Du HN, Du LL, du Toit A, Duan SB, Duan X, Duarte SP, Dubrovska A, Dunlop EA, Dupont N, Durán RV, Dwarakanath BS, Dyshlovoy SA, Ebrahimi-Fakhari D, Eckhart L, Edelstein CL, Efferth T, Eftekharpour E, Eichinger L, Eid N, Eisenberg T, Eissa NT, Eissa S, Ejarque M, El Andaloussi A, El-Hage N, El-Naggar S, Eleuteri AM, El-Shafey ES, Elgendy M, Eliopoulos AG, Elizalde MM, Elks PM, Elsasser HP, Elsherbiny ES, Emerling BM, Emre NCT, Eng CH, Engedal N, Engelbrecht AM, Engelsen AST, Enserink JM, Escalante R, Esclatine A, Escobar-Henriques M, Eskelinen EL, Espert L, Eusebio MO, Fabrias G, Fabrizi C, Facchiano A, Facchiano F, Fadeel B, Fader C, Faesen AC, Fairlie WD, Falcó A, Falkenburger BH, Fan D, Fan J, Fan Y, Fang EF, Fang Y, Fang Y, Fanto M, Farfel-Becker T, Faure M, Fazeli G, Fedele AO, Feldman AM, Feng D, Feng J, Feng L, Feng Y, Feng Y, Feng W, Fenz Araujo T, Ferguson TA, Fernández ÁF, Fernandez-Checa JC, Fernández-Veledo S, Fernie AR, Ferrante AW Jr, Ferraresi A, Ferrari MF, Ferreira JCB, Ferro-Novick S, Figueras A, Filadi R, Filigheddu N, Filippi-Chiela E, Filomeni G, Fimia GM, Fineschi V, Finetti F, Finkbeiner S, Fisher EA, Fisher PB, Flamigni F, Fliesler SJ, Flo TH, Florance I, Florey O, Florio T, Fodor E, Follo C, Fon EA, Forlino A, Fornai F, Fortini P, Fracassi A, Fraldi A, Franco B, Franco R, Franconi F, Frankel LB, Friedman SL, Fröhlich LF, Frühbeck G, Fuentes JM, Fujiki Y, Fujita N, Fujiwara Y, Fukuda M, Fulda S, Furic L, Furuya N, Fusco C, Gack MU, Gaffke L, Galadari S, Galasso A, Galindo MF, Gallolu Kankanamalage S, Galluzzi L, Galy V, Gammoh N, Gan B, Ganley IG, Gao F, Gao H, Gao M, Gao P, Gao SJ, Gao W, Gao X, Garcera A, Garcia MN, Garcia VE, García-Del Portillo F, Garcia-Escudero V, Garcia-Garcia A, Garcia-Macia M, García-Moreno D, Garcia-Ruiz C, García-Sanz P, Garg AD, Gargini R, Garofalo T, Garry RF, Gassen NC, Gatica D, Ge L, Ge W, Geiss-Friedlander R, Gelfi C, Genschik P, Gentle IE, Gerbino V, Gerhardt C, Germain K, Germain M, Gewirtz DA, Ghasemipour Afshar E, Ghavami S, Ghigo A, Ghosh M, Giamas G, Giampietri C, Giatromanolaki A, Gibson GE, Gibson SB, Ginet V, Giniger E, Giorgi C, Girao H, Girardin SE, Giridharan M, Giuliano S, Giulivi C, Giuriato S, Giustiniani J, Gluschko A, Goder V, Goginashvili A, Golab J, Goldstone DC, Golebiewska A, Gomes LR, Gomez R, Gómez-Sánchez R, Gomez-Puerto MC, Gomez-Sintes R, Gong Q, Goni FM, González-Gallego J, Gonzalez-Hernandez T, Gonzalez-Polo RA, Gonzalez-Reyes JA, González-Rodríguez P, Goping IS, Gorbatyuk MS, Gorbunov NV, Görgülü K, Gorojod RM, Gorski SM, Goruppi S, Gotor C, Gottlieb RA, Gozes I, Gozuacik D, Graef M, Gräler MH, Granatiero V, Grasso D, Gray JP, Green DR, Greenhough A, Gregory SL, Griffin EF, Grinstaff MW, Gros F, Grose C, Gross AS, Gruber F, Grumati P, Grune T, Gu X, Guan JL, Guardia CM, Guda K, Guerra F, Guerri C, Guha P, Guillén C, Gujar S, Gukovskaya A, Gukovsky I, Gunst J, Günther A, Guntur AR, Guo C, Guo C, Guo H, Guo LW, Guo M, Gupta P, Gupta SK, Gupta S, Gupta VB, Gupta V, Gustafsson AB, Gutterman DD, H B R, Haapasalo A, Haber JE, Hać A, Hadano S, Hafrén AJ, Haidar M, Hall BS, Halldén G, Hamacher-Brady A, Hamann A, Hamasaki M, Han W, Hansen M, Hanson PI, Hao Z, Harada M, Harhaji-Trajkovic L, Hariharan N, Haroon N, Harris J, Hasegawa T, Hasima Nagoor N, Haspel JA, Haucke V, Hawkins WD, Hay BA, Haynes CM, Hayrabedyan SB, Hays TS, He C, He Q, He RR, He YW, He YY, Heakal Y, Heberle AM, Hejtmancik JF, Helgason GV, Henkel V, Herb M, Hergovich A, Herman-Antosiewicz A, Hernández A, Hernandez C, Hernandez-Diaz S, Hernandez-Gea V, Herpin A, Herreros J, Hervás JH, Hesselson D, Hetz C, Heussler VT, Higuchi Y, Hilfiker S, Hill JA, Hlavacek WS, Ho EA, Ho IHT, Ho PW, Ho SL, Ho WY, Hobbs GA, Hochstrasser M, Hoet PHM, Hofius D, Hofman P, Höhn A, Holmberg CI, Hombrebueno JR, Yi-Ren Hong CH, Hooper LV, Hoppe T, Horos R, Hoshida Y, Hsin IL, Hsu HY, Hu B, Hu D, Hu LF, Hu MC, Hu R, Hu W, Hu YC, Hu ZW, Hua F, Hua J, Hua Y, Huan C, Huang C, Huang C, Huang C, Huang C, Huang H, Huang K, Huang MLH, Huang R, Huang S, Huang T, Huang X, Huang YJ, Huber TB, Hubert V, Hubner CA, Hughes SM, Hughes WE, Humbert M, Hummer G, Hurley JH, Hussain S, Hussain S, Hussey PJ, Hutabarat M, Hwang HY, Hwang S, Ieni A, Ikeda F, Imagawa Y, Imai Y, Imbriano C, Imoto M, Inman DM, Inoki K, Iovanna J, Iozzo RV, Ippolito G, Irazoqui JE, Iribarren P, Ishaq M, Ishikawa M, Ishimwe N, Isidoro C, Ismail N, Issazadeh-Navikas S, Itakura E, Ito D, Ivankovic D, Ivanova S, Iyer AKV, Izquierdo JM, Izumi M, Jäättelä M, Jabir MS, Jackson WT, Jacobo-Herrera N, Jacomin AC, Jacquin E, Jadiya P, Jaeschke H, Jagannath C, Jakobi AJ, Jakobsson J, Janji B, Jansen-Dürr P, Jansson PJ, Jantsch J, Januszewski S, Jassey A, Jean S, Jeltsch-David H, Jendelova P, Jenny A, Jensen TE, Jessen N, Jewell JL, Ji J, Jia L, Jia R, Jiang L, Jiang Q, Jiang R, Jiang T, Jiang X, Jiang Y, Jimenez-Sanchez M, Jin EJ, Jin F, Jin H, Jin L, Jin L, Jin M, Jin S, Jo EK, Joffre C, Johansen T, Johnson GVW, Johnston SA, Jokitalo E, Jolly MK, Joosten LAB, Jordan J, Joseph B, Ju D, Ju JS, Ju J, Juárez E, Judith D, Juhász G, Jun Y, Jung CH, Jung SC, Jung YK, Jungbluth H, Jungverdorben J, Just S, Kaarniranta K, Kaasik A, Kabuta T, Kaganovich D, Kahana A, Kain R, Kajimura S, Kalamvoki M, Kalia M, Kalinowski DS, Kaludercic N, Kalvari I, Kaminska J, Kaminskyy VO, Kanamori H, Kanasaki K, Kang C, Kang R, Kang SS, Kaniyappan S, Kanki T, Kanneganti TD, Kanthasamy AG, Kanthasamy A, Kantorow M, Kapuy O, Karamouzis MV, Karim MR, Karmakar P, Katare RG, Kato M, Kaufmann SHE, Kauppinen A, Kaushal GP, Kaushik S, Kawasaki K, Kazan K, Ke PY, Keating DJ, Keber U, Kehrl JH, Keller KE, Keller CW, Kemper JK, Kenific CM, Kepp O, Kermorgant S, Kern A, Ketteler R, Keulers TG, Khalfin B, Khalil H, Khambu B, Khan SY, Khandelwal VKM, Khandia R, Kho W, Khobrekar NV, Khuansuwan S, Khundadze M, Killackey SA, Kim D, Kim DR, Kim DH, Kim DE, Kim EY, Kim EK, Kim HR, Kim HS, Hyung-Ryong Kim, Kim JH, Kim JK, Kim JH, Kim J, Kim JH, Kim KI, Kim PK, Kim SJ, Kimball SR, Kimchi A, Kimmelman AC, Kimura T, King MA, Kinghorn KJ, Kinsey CG, Kirkin V, Kirshenbaum LA, Kiselev SL, Kishi S, Kitamoto K, Kitaoka Y, Kitazato K, Kitsis RN, Kittler JT, Kjaerulff O, Klein PS, Klopstock T, Klucken J, Knævelsrud H, Knorr RL, Ko BCB, Ko F, Ko JL, Kobayashi H, Kobayashi S, Koch I, Koch JC, Koenig U, Kögel D, Koh YH, Koike M, Kohlwein SD, Kocaturk NM, Komatsu M, König J, Kono T, Kopp BT, Korcsmaros T, Korkmaz G, Korolchuk VI, Korsnes MS, Koskela A, Kota J, Kotake Y, Kotler ML, Kou Y, Koukourakis MI, Koustas E, Kovacs AL, Kovács T, Koya D, Kozako T, Kraft C, Krainc D, Krämer H, Krasnodembskaya AD, Kretz-Remy C, Kroemer G, Ktistakis NT, Kuchitsu K, Kuenen S, Kuerschner L, Kukar T, Kumar A, Kumar A, Kumar D, Kumar D, Kumar S, Kume S, Kumsta C, Kundu CN, Kundu M, Kunnumakkara AB, Kurgan L, Kutateladze TG, Kutlu O, Kwak S, Kwon HJ, Kwon TK, Kwon YT, Kyrmizi I, La Spada A, Labonté P, Ladoire S, Laface I, Lafont F, Lagace DC, Lahiri V, Lai Z, Laird AS, Lakkaraju A, Lamark T, Lan SH, Landajuela A, Lane DJR, Lane JD, Lang CH, Lange C, Langel Ü, Langer R, Lapaquette P, Laporte J, LaRusso NF, Lastres-Becker I, Lau WCY, Laurie GW, Lavandero S, Law BYK, Law HK, Layfield R, Le W, Le Stunff H, Leary AY, Lebrun JJ, Leck LYW, Leduc-Gaudet JP, Lee C, Lee CP, Lee DH, Lee EB, Lee EF, Lee GM, Lee HJ, Lee HK, Lee JM, Lee JS, Lee JA, Lee JY, Lee JH, Lee M, Lee MG, Lee MJ, Lee MS, Lee SY, Lee SJ, Lee SY, Lee SB, Lee WH, Lee YR, Lee YH, Lee Y, Lefebvre C, Legouis R, Lei YL, Lei Y, Leikin S, Leitinger G, Lemus L, Leng S, Lenoir O, Lenz G, Lenz HJ, Lenzi P, León Y, Leopoldino AM, Leschczyk C, Leskelä S, Letellier E, Leung CT, Leung PS, Leventhal JS, Levine B, Lewis PA, Ley K, Li B, Li DQ, Li J, Li J, Li J, Li K, Li L, Li M, Li M, Li M, Li M, Li M, Li PL, Li MQ, Li Q, Li S, Li T, Li W, Li W, Li X, Li YP, Li Y, Li Z, Li Z, Li Z, Lian J, Liang C, Liang Q, Liang W, Liang Y, Liang Y, Liao G, Liao L, Liao M, Liao YF, Librizzi M, Lie PPY, Lilly MA, Lim HJ, Lima TRR, Limana F, Lin C, Lin CW, Lin DS, Lin FC, Lin JD, Lin KM, Lin KH, Lin LT, Lin PH, Lin Q, Lin S, Lin SJ, Lin W, Lin X, Lin YX, Lin YS, Linden R, Lindner P, Ling SC, Lingor P, Linnemann AK, Liou YC, Lipinski MM, Lipovšek S, Lira VA, Lisiak N, Liton PB, Liu C, Liu CH, Liu CF, Liu CH, Liu F, Liu H, Liu HS, Liu HF, Liu H, Liu J, Liu J, Liu J, Liu L, Liu L, Liu M, Liu Q, Liu W, Liu W, Liu XH, Liu X, Liu X, Liu X, Liu X, Liu Y, Liu Y, Liu Y, Liu Y, Liu Y, Livingston JA, Lizard G, Lizcano JM, Ljubojevic-Holzer S, LLeonart ME, Llobet-Navàs D, Llorente A, Lo CH, Lobato-Márquez D, Long Q, Long YC, Loos B, Loos JA, López MG, López-Doménech G, López-Guerrero JA, López-Jiménez AT, López-Pérez Ó, López-Valero I, Lorenowicz MJ, Lorente M, Lorincz P, Lossi L, Lotersztajn S, Lovat PE, Lovell JF, Lovy A, Lőw P, Lu G, Lu H, Lu JH, Lu JJ, Lu M, Lu S, Luciani A, Lucocq JM, Ludovico P, Luftig MA, Luhr M, Luis-Ravelo D, Lum JJ, Luna-Dulcey L, Lund AH, Lund VK, Lünemann JD, Lüningschrör P, Luo H, Luo R, Luo S, Luo Z, Luparello C, Lüscher B, Luu L, Lyakhovich A, Lyamzaev KG, Lystad AH, Lytvynchuk L, Ma AC, Ma C, Ma M, Ma NF, Ma QH, Ma X, Ma Y, Ma Z, MacDougald OA, Macian F, MacIntosh GC, MacKeigan JP, Macleod KF, Maday S, Madeo F, Madesh M, Madl T, Madrigal-Matute J, Maeda A, Maejima Y, Magarinos M, Mahavadi P, Maiani E, Maiese K, Maiti P, Maiuri MC, Majello B, Major MB, Makareeva E, Malik F, Mallilankaraman K, Malorni W, Maloyan A, Mammadova N, Man GCW, Manai F, Mancias JD, Mandelkow EM, Mandell MA, Manfredi AA, Manjili MH, Manjithaya R, Manque P, Manshian BB, Manzano R, Manzoni C, Mao K, Marchese C, Marchetti S, Marconi AM, Marcucci F, Mardente S, Mareninova OA, Margeta M, Mari M, Marinelli S, Marinelli O, Mariño G, Mariotto S, Marshall RS, Marten MR, Martens S, Martin APJ, Martin KR, Martin S, Martin S, Martín-Segura A, Martín-Acebes MA, Martin-Burriel I, Martin-Rincon M, Martin-Sanz P, Martina JA, Martinet W, Martinez A, Martinez A, Martinez J, Martinez Velazquez M, Martinez-Lopez N, Martinez-Vicente M, Martins DO, Martins JO, Martins WK, Martins-Marques T, Marzetti E, Masaldan S, Masclaux-Daubresse C, Mashek DG, Massa V, Massieu L, Masson GR, Masuelli L, Masyuk AI, Masyuk TV, Matarrese P, Matheu A, Matoba S, Matsuzaki S, Mattar P, Matte A, Mattoscio D, Mauriz JL, Mauthe M, Mauvezin C, Maverakis E, Maycotte P, Mayer J, Mazzoccoli G, Mazzoni C, Mazzulli JR, McCarty N, McDonald C, McGill MR, McKenna SL, McLaughlin B, McLoughlin F, McNiven MA, McWilliams TG, Mechta-Grigoriou F, Medeiros TC, Medina DL, Megeney LA, Megyeri K, Mehrpour M, Mehta JL, Meijer AJ, Meijer AH, Mejlvang J, Meléndez A, Melk A, Memisoglu G, Mendes AF, Meng D, Meng F, Meng T, Menna-Barreto R, Menon MB, Mercer C, Mercier AE, Mergny JL, Merighi A, Merkley SD, Merla G, Meske V, Mestre AC, Metur SP, Meyer C, Meyer H, Mi W, Mialet-Perez J, Miao J, Micale L, Miki Y, Milan E, Milczarek M, Miller DL, Miller SI, Miller S, Millward SW, Milosevic I, Minina EA, Mirzaei H, Mirzaei HR, Mirzaei M, Mishra A, Mishra N, Mishra PK, Misirkic Marjanovic M, Misasi R, Misra A, Misso G, Mitchell C, Mitou G, Miura T, Miyamoto S, Miyazaki M, Miyazaki M, Miyazaki T, Miyazawa K, Mizushima N, Mogensen TH, Mograbi B, Mohammadinejad R, Mohamud Y, Mohanty A, Mohapatra S, Möhlmann T, Mohmmed A, Moles A, Moley KH, Molinari M, Mollace V, Møller AB, Mollereau B, Mollinedo F, Montagna C, Monteiro MJ, Montella A, Montes LR, Montico B, Mony VK, Monzio Compagnoni G, Moore MN, Moosavi MA, Mora AL, Mora M, Morales-Alamo D, Moratalla R, Moreira PI, Morelli E, Moreno S, Moreno-Blas D, Moresi V, Morga B, Morgan AH, Morin F, Morishita H, Moritz OL, Moriyama M, Moriyasu Y, Morleo M, Morselli E, Moruno-Manchon JF, Moscat J, Mostowy S, Motori E, Moura AF, Moustaid-Moussa N, Mrakovcic M, Muciño-Hernández G, Mukherjee A, Mukhopadhyay S, Mulcahy Levy JM, Mulero V, Muller S, Münch C, Munjal A, Munoz-Canoves P, Muñoz-Galdeano T, Münz C, Murakawa T, Muratori C, Murphy BM, Murphy JP, Murthy A, Myöhänen TT, Mysorekar IU, Mytych J, Nabavi SM, Nabissi M, Nagy P, Nah J, Nahimana A, Nakagawa I, Nakamura K, Nakatogawa H, Nandi SS, Nanjundan M, Nanni M, Napolitano G, Nardacci R, Narita M, Nassif M, Nathan I, Natsumeda M, Naude RJ, Naumann C, Naveiras O, Navid F, Nawrocki ST, Nazarko TY, Nazio F, Negoita F, Neill T, Neisch AL, Neri LM, Netea MG, Neubert P, Neufeld TP, Neumann D, Neutzner A, Newton PT, Ney PA, Nezis IP, Ng CCW, Ng TB, Nguyen HTT, Nguyen LT, Ni HM, Ní Cheallaigh C, Ni Z, Nicolao MC, Nicoli F, Nieto-Diaz M, Nilsson P, Ning S, Niranjan R, Nishimune H, Niso-Santano M, Nixon RA, Nobili A, Nobrega C, Noda T, Nogueira-Recalde U, Nolan TM, Nombela I, Novak I, Novoa B, Nozawa T, Nukina N, Nussbaum-Krammer C, Nylandsted J, O'Donovan TR, O'Leary SM, O'Rourke EJ, O'Sullivan MP, O'Sullivan TE, Oddo S, Oehme I, Ogawa M, Ogier-Denis E, Ogmundsdottir MH, Ogretmen B, Oh GT, Oh SH, Oh YJ, Ohama T, Ohashi Y, Ohmuraya M, Oikonomou V, Ojha R, Okamoto K, Okazawa H, Oku M, Oliván S, Oliveira JMA, Ollmann M, Olzmann JA, Omari S, Omary MB, Önal G, Ondrej M, Ong SB, Ong SG, Onnis A, Orellana JA, Orellana-Muñoz S, Ortega-Villaizan MDM, Ortiz-Gonzalez XR, Ortona E, Osiewacz HD, Osman AK, Osta R, Otegui MS, Otsu K, Ott C, Ottobrini L, Ou JJ, Outeiro TF, Oynebraten I, Ozturk M, Pagès G, Pahari S, Pajares M, Pajvani UB, Pal R, Paladino S, Pallet N, Palmieri M, Palmisano G, Palumbo C, Pampaloni F, Pan L, Pan Q, Pan W, Pan X, Panasyuk G, Pandey R, Pandey UB, Pandya V, Paneni F, Pang SY, Panzarini E, Papademetrio DL, Papaleo E, Papinski D, Papp D, Park EC, Park HT, Park JM, Park JI, Park JT, Park J, Park SC, Park SY, Parola AH, Parys JB, Pasquier A, Pasquier B, Passos JF, Pastore N, Patel HH, Patschan D, Pattingre S, Pedraza-Alva G, Pedraza-Chaverri J, Pedrozo Z, Pei G, Pei J, Peled-Zehavi H, Pellegrini JM, Pelletier J, Peñalva MA, Peng D, Peng Y, Penna F, Pennuto M, Pentimalli F, Pereira CM, Pereira GJS, Pereira LC, Pereira de Almeida L, Perera ND, Pérez-Lara Á, Perez-Oliva AB, Pérez-Pérez ME, Periyasamy P, Perl A, Perrotta C, Perrotta I, Pestell RG, Petersen M, Petrache I, Petrovski G, Pfirrmann T, Pfister AS, Philips JA, Pi H, Picca A, Pickrell AM, Picot S, Pierantoni GM, Pierdominici M, Pierre P, Pierrefite-Carle V, Pierzynowska K, Pietrocola F, Pietruczuk M, Pignata C, Pimentel-Muiños FX, Pinar M, Pinheiro RO, Pinkas-Kramarski R, Pinton P, Pircs K, Piya S, Pizzo P, Plantinga TS, Platta HW, Plaza-Zabala A, Plomann M, Plotnikov EY, Plun-Favreau H, Pluta R, Pocock R, Pöggeler S, Pohl C, Poirot M, Poletti A, Ponpuak M, Popelka H, Popova B, Porta H, Porte Alcon S, Portilla-Fernandez E, Post M, Potts MB, Poulton J, Powers T, Prahlad V, Prajsnar TK, Praticò D, Prencipe R, Priault M, Proikas-Cezanne T, Promponas VJ, Proud CG, Puertollano R, Puglielli L, Pulinilkunnil T, Puri D, Puri R, Puyal J, Qi X, Qi Y, Qian W, Qiang L, Qiu Y, Quadrilatero J, Quarleri J, Raben N, Rabinowich H, Ragona D, Ragusa MJ, Rahimi N, Rahmati M, Raia V, Raimundo N, Rajasekaran NS, Ramachandra Rao S, Rami A, Ramírez-Pardo I, Ramsden DB, Randow F, Rangarajan PN, Ranieri D, Rao H, Rao L, Rao R, Rathore S, Ratnayaka JA, Ratovitski EA, Ravanan P, Ravegnini G, Ray SK, Razani B, Rebecca V, Reggiori F, Régnier-Vigouroux A, Reichert AS, Reigada D, Reiling JH, Rein T, Reipert S, Rekha RS, Ren H, Ren J, Ren W, Renault T, Renga G, Reue K, Rewitz K, Ribeiro de Andrade Ramos B, Riazuddin SA, Ribeiro-Rodrigues TM, Ricci JE, Ricci R, Riccio V, Richardson DR, Rikihisa Y, Risbud MV, Risueño RM, Ritis K, Rizza S, Rizzuto R, Roberts HC, Roberts LD, Robinson KJ, Roccheri MC, Rocchi S, Rodney GG, Rodrigues T, Rodrigues Silva VR, Rodriguez A, Rodriguez-Barrueco R, Rodriguez-Henche N, Rodriguez-Rocha H, Roelofs J, Rogers RS, Rogov VV, Rojo AI, Rolka K, Romanello V, Romani L, Romano A, Romano PS, Romeo-Guitart D, Romero LC, Romero M, Roney JC, Rongo C, Roperto S, Rosenfeldt MT, Rosenstiel P, Rosenwald AG, Roth KA, Roth L, Roth S, Rouschop KMA, Roussel BD, Roux S, Rovere-Querini P, Roy A, Rozieres A, Ruano D, Rubinsztein DC, Rubtsova MP, Ruckdeschel K, Ruckenstuhl C, Rudolf E, Rudolf R, Ruggieri A, Ruparelia AA, Rusmini P, Russell RR, Russo GL, Russo M, Russo R, Ryabaya OO, Ryan KM, Ryu KY, Sabater-Arcis M, Sachdev U, Sacher M, Sachse C, Sadhu A, Sadoshima J, Safren N, Saftig P, Sagona AP, Sahay G, Sahebkar A, Sahin M, Sahin O, Sahni S, Saito N, Saito S, Saito T, Sakai R, Sakai Y, Sakamaki JI, Saksela K, Salazar G, Salazar-Degracia A, Salekdeh GH, Saluja AK, Sampaio-Marques B, Sanchez MC, Sanchez-Alcazar JA, Sanchez-Vera V, Sancho-Shimizu V, Sanderson JT, Sandri M, Santaguida S, Santambrogio L, Santana MM, Santoni G, Sanz A, Sanz P, Saran S, Sardiello M, Sargeant TJ, Sarin A, Sarkar C, Sarkar S, Sarrias MR, Sarkar S, Sarmah DT, Sarparanta J, Sathyanarayan A, Sathyanarayanan R, Scaglione KM, Scatozza F, Schaefer L, Schafer ZT, Schaible UE, Schapira AHV, Scharl M, Schatzl HM, Schein CH, Scheper W, Scheuring D, Schiaffino MV, Schiappacassi M, Schindl R, Schlattner U, Schmidt O, Schmitt R, Schmidt SD, Schmitz I, Schmukler E, Schneider A, Schneider BE, Schober R, Schoijet AC, Schott MB, Schramm M, Schröder B, Schuh K, Schüller C, Schulze RJ, Schürmanns L, Schwamborn JC, Schwarten M, Scialo F, Sciarretta S, Scott MJ, Scotto KW, Scovassi AI, Scrima A, Scrivo A, Sebastian D, Sebti S, Sedej S, Segatori L, Segev N, Seglen PO, Seiliez I, Seki E, Selleck SB, Sellke FW, Selsby JT, Sendtner M, Senturk S, Seranova E, Sergi C, Serra-Moreno R, Sesaki H, Settembre C, Setty SRG, Sgarbi G, Sha O, Shacka JJ, Shah JA, Shang D, Shao C, Shao F, Sharbati S, Sharkey LM, Sharma D, Sharma G, Sharma K, Sharma P, Sharma S, Shen HM, Shen H, Shen J, Shen M, Shen W, Shen Z, Sheng R, Sheng Z, Sheng ZH, Shi J, Shi X, Shi YH, Shiba-Fukushima K, Shieh JJ, Shimada Y, Shimizu S, Shimozawa M, Shintani T, Shoemaker CJ, Shojaei S, Shoji I, Shravage BV, Shridhar V, Shu CW, Shu HB, Shui K, Shukla AK, Shutt TE, Sica V, Siddiqui A, Sierra A, Sierra-Torre V, Signorelli S, Sil P, Silva BJA, Silva JD, Silva-Pavez E, Silvente-Poirot S, Simmonds RE, Simon AK, Simon HU, Simons M, Singh A, Singh LP, Singh R, Singh SV, Singh SK, Singh SB, Singh S, Singh SP, Sinha D, Sinha RA, Sinha S, Sirko A, Sirohi K, Sivridis EL, Skendros P, Skirycz A, Slaninová I, Smaili SS, Smertenko A, Smith MD, Soenen SJ, Sohn EJ, Sok SPM, Solaini G, Soldati T, Soleimanpour SA, Soler RM, Solovchenko A, Somarelli JA, Sonawane A, Song F, Song HK, Song JX, Song K, Song Z, Soria LR, Sorice M, Soukas AA, Soukup SF, Sousa D, Sousa N, Spagnuolo PA, Spector SA, Srinivas Bharath MM, St Clair D, Stagni V, Staiano L, Stalnecker CA, Stankov MV, Stathopulos PB, Stefan K, Stefan SM, Stefanis L, Steffan JS, Steinkasserer A, Stenmark H, Sterneckert J, Stevens C, Stoka V, Storch S, Stork B, Strappazzon F, Strohecker AM, Stupack DG, Su H, Su LY, Su L, Suarez-Fontes AM, Subauste CS, Subbian S, Subirada PV, Sudhandiran G, Sue CM, Sui X, Summers C, Sun G, Sun J, Sun K, Sun MX, Sun Q, Sun Y, Sun Z, Sunahara KKS, Sundberg E, Susztak K, Sutovsky P, Suzuki H, Sweeney G, Symons JD, Sze SCW, Szewczyk NJ, Tabęcka-Łonczynska A, Tabolacci C, Tacke F, Taegtmeyer H, Tafani M, Tagaya M, Tai H, Tait SWG, Takahashi Y, Takats S, Talwar P, Tam C, Tam SY, Tampellini D, Tamura A, Tan CT, Tan EK, Tan YQ, Tanaka M, Tanaka M, Tang D, Tang J, Tang TS, Tanida I, Tao Z, Taouis M, Tatenhorst L, Tavernarakis N, Taylor A, Taylor GA, Taylor JM, Tchetina E, Tee AR, Tegeder I, Teis D, Teixeira N, Teixeira-Clerc F, Tekirdag KA, Tencomnao T, Tenreiro S, Tepikin AV, Testillano PS, Tettamanti G, Tharaux PL, Thedieck K, Thekkinghat AA, Thellung S, Thinwa JW, Thirumalaikumar VP, Thomas SM, Thomes PG, Thorburn A, Thukral L, Thum T, Thumm M, Tian L, Tichy A, Till A, Timmerman V, Titorenko VI, Todi SV, Todorova K, Toivonen JM, Tomaipitinca L, Tomar D, Tomas-Zapico C, Tomić S, Tong BC, Tong C, Tong X, Tooze SA, Torgersen ML, Torii S, Torres-López L, Torriglia A, Towers CG, Towns R, Toyokuni S, Trajkovic V, Tramontano D, Tran QG, Travassos LH, Trelford CB, Tremel S, Trougakos IP, Tsao BP, Tschan MP, Tse HF, Tse TF, Tsugawa H, Tsvetkov AS, Tumbarello DA, Tumtas Y, Tuñón MJ, Turcotte S, Turk B, Turk V, Turner BJ, Tuxworth RI, Tyler JK, Tyutereva EV, Uchiyama Y, Ugun-Klusek A, Uhlig HH, Ułamek-Kozioł M, Ulasov IV, Umekawa M, Ungermann C, Unno R, Urbe S, Uribe-Carretero E, Üstün S, Uversky VN, Vaccari T, Vaccaro MI, Vahsen BF, Vakifahmetoglu-Norberg H, Valdor R, Valente MJ, Valko A, Vallee RB, Valverde AM, Van den Berghe G, van der Veen S, Van Kaer L, van Loosdregt J, van Wijk SJL, Vandenberghe W, Vanhorebeek I, Vannier-Santos MA, Vannini N, Vanrell MC, Vantaggiato C, Varano G, Varela-Nieto I, Varga M, Vasconcelos MH, Vats S, Vavvas DG, Vega-Naredo I, Vega-Rubin-de-Celis S, Velasco G, Velázquez AP, Vellai T, Vellenga E, Velotti F, Verdier M, Verginis P, Vergne I, Verkade P, Verma M, Verstreken P, Vervliet T, Vervoorts J, Vessoni AT, Victor VM, Vidal M, Vidoni C, Vieira OV, Vierstra RD, Viganó S, Vihinen H, Vijayan V, Vila M, Vilar M, Villalba JM, Villalobo A, Villarejo-Zori B, Villarroya F, Villarroya J, Vincent O, Vindis C, Viret C, Viscomi MT, Visnjic D, Vitale I, Vocadlo DJ, Voitsekhovskaja OV, Volonté C, Volta M, Vomero M, Von Haefen C, Vooijs MA, Voos W, Vucicevic L, Wade-Martins R, Waguri S, Waite KA, Wakatsuki S, Walker DW, Walker MJ, Walker SA, Walter J, Wandosell FG, Wang B, Wang CY, Wang C, Wang C, Wang C, Wang CY, Wang D, Wang F, Wang F, Wang F, Wang G, Wang H, Wang H, Wang H, Wang HG, Wang J, Wang J, Wang J, Wang J, Wang K, Wang L, Wang L, Wang MH, Wang M, Wang N, Wang P, Wang P, Wang P, Wang P, Wang QJ, Wang Q, Wang QK, Wang QA, Wang WT, Wang W, Wang X, Wang X, Wang Y, Wang Y, Wang Y, Wang YY, Wang Y, Wang Y, Wang Y, Wang Y, Wang Z, Wang Z, Wang Z, Warnes G, Warnsmann V, Watada H, Watanabe E, Watchon M, Wawrzyńska A, Weaver TE, Wegrzyn G, Wehman AM, Wei H, Wei L, Wei T, Wei Y, Weiergräber OH, Weihl CC, Weindl G, Weiskirchen R, Wells A, Wen RH, Wen X, Werner A, Weykopf B, Wheatley SP, Whitton JL, Whitworth AJ, Wiktorska K, Wildenberg ME, Wileman T, Wilkinson S, Willbold D, Williams B, Williams RSB, Williams RL, Williamson PR, Wilson RA, Winner B, Winsor NJ, Witkin SS, Wodrich H, Woehlbier U, Wollert T, Wong E, Wong JH, Wong RW, Wong VKW, Wong WW, Wu AG, Wu C, Wu J, Wu J, Wu KK, Wu M, Wu SY, Wu S, Wu SY, Wu S, Wu WKK, Wu X, Wu X, Wu YW, Wu Y, Xavier RJ, Xia H, Xia L, Xia Z, Xiang G, Xiang J, Xiang M, Xiang W, Xiao B, Xiao G, Xiao H, Xiao HT, Xiao J, Xiao L, Xiao S, Xiao Y, Xie B, Xie CM, Xie M, Xie Y, Xie Z, Xie Z, Xilouri M, Xu C, Xu E, Xu H, Xu J, Xu J, Xu L, Xu WW, Xu X, Xue Y, Yakhine-Diop SMS, Yamaguchi M, Yamaguchi O, Yamamoto A, Yamashina S, Yan S, Yan SJ, Yan Z, Yanagi Y, Yang C, Yang DS, Yang H, Yang HT, Yang H, Yang JM, Yang J, Yang J, Yang L, Yang L, Yang M, Yang PM, Yang Q, Yang S, Yang S, Yang SF, Yang W, Yang WY, Yang X, Yang X, Yang Y, Yang Y, Yao H, Yao S, Yao X, Yao YG, Yao YM, Yasui T, Yazdankhah M, Yen PM, Yi C, Yin XM, Yin Y, Yin Z, Yin Z, Ying M, Ying Z, Yip CK, Yiu SPT, Yoo YH, Yoshida K, Yoshii SR, Yoshimori T, Yousefi B, Yu B, Yu H, Yu J, Yu J, Yu L, Yu ML, Yu SW, Yu VC, Yu WH, Yu Z, Yu Z, Yuan J, Yuan LQ, Yuan S, Yuan SF, Yuan Y, Yuan Z, Yue J, Yue Z, Yun J, Yung RL, Zacks DN, Zaffagnini G, Zambelli VO, Zanella I, Zang QS, Zanivan S, Zappavigna S, Zaragoza P, Zarbalis KS, Zarebkohan A, Zarrouk A, Zeitlin SO, Zeng J, Zeng JD, Žerovnik E, Zhan L, Zhang B, Zhang DD, Zhang H, Zhang H, Zhang H, Zhang H, Zhang H, Zhang H, Zhang H, Zhang HL, Zhang J, Zhang J, Zhang JP, Zhang KYB, Zhang LW, Zhang L, Zhang L, Zhang L, Zhang L, Zhang M, Zhang P, Zhang S, Zhang W, Zhang X, Zhang XW, Zhang X, Zhang X, Zhang X, Zhang X, Zhang XD, Zhang Y, Zhang Y, Zhang Y, Zhang YD, Zhang Y, Zhang YY, Zhang Y, Zhang Z, Zhang Z, Zhang Z, Zhang Z, Zhang Z, Zhang Z, Zhao H, Zhao L, Zhao S, Zhao T, Zhao XF, Zhao Y, Zhao Y, Zhao Y, Zhao Y, Zheng G, Zheng K, Zheng L, Zheng S, Zheng XL, Zheng Y, Zheng ZG, Zhivotovsky B, Zhong Q, Zhou A, Zhou B, Zhou C, Zhou G, Zhou H, Zhou H, Zhou H, Zhou J, Zhou J, Zhou J, Zhou J, Zhou K, Zhou R, Zhou XJ, Zhou Y, Zhou Y, Zhou Y, Zhou ZY, Zhou Z, Zhu B, Zhu C, Zhu GQ, Zhu H, Zhu H, Zhu H, Zhu WG, Zhu Y, Zhu Y, Zhuang H, Zhuang X, Zientara-Rytter K, Zimmermann CM, Ziviani E, Zoladek T, Zong WX, Zorov DB, Zorzano A, Zou W, Zou Z, Zou Z, Zuryn S, Zwerschke W, Brand-Saberi B, Dong XC, Kenchappa CS, Li Z, Lin Y, Oshima S, Rong Y, Sluimer JC, Stallings CL, Tong CK. Guidelines for the use and interpretation of assays for monitoring autophagy (4th edition)1. Autophagy. 2021 Jan;17(1):1-382. Epub 2021 Feb 8. PMID: 33634751; PMCID: PMC7996087.
Haack TB, Ignatius E, Calvo-Garrido J, Iuso A, Isohanni P, Maffezzini C, Lönnqvist T, Suomalainen A, Gorza M, Kremer LS, Graf E, Hartig M, Berutti R, Paucar M, Svenningsson P, Stranneheim H, Brandberg G, Wedell A, Kurian MA, Hayflick SA, Venco P, Tiranti V, Strom TM, Dichgans M, Horvath R, Holinski-Feder E, Freyer C, Meitinger T, Prokisch H, Senderek J, Wredenberg A, Carroll CJ, Klopstock T. Absence of the Autophagy Adaptor SQSTM1/p62 Causes Childhood-Onset Neurodegeneration with Ataxia, Dystonia, and Gaze Palsy. Am J Hum Genet. 2016 Sep 1;99(3):735-743. Epub 2016 Aug 18. PMID: 27545679; PMCID: PMC5010644. DOI: https://doi.org/10.1016/j.ajhg.2016.06.026
Okatsu K, Saisho K, Shimanuki M, Nakada K, Shitara H, Sou YS, Kimura M, Sato S, Hattori N, Komatsu M, Tanaka K, Matsuda N. p62/SQSTM1 cooperates with Parkin for perinuclear clustering of depolarized mitochondria. Genes Cells. 2010 Aug;15(8):887-900. Epub 2010 Jul 2. PMID: 20604804; PMCID: PMC2970908. DOI: https://doi.org/10.1111/j.1365-2443.2010.01426.x
Liu Y, Levine B. Autosis and autophagic cell death: the dark side of autophagy. Cell Death Differ. 2015 Mar;22(3):367-76. Epub 2014 Sep 26. PMID: 25257169; PMCID: PMC4326571. DOI: https://doi.org/10.1038/cdd.2014.143
De Palma C, Morisi F, Cheli S, Pambianco S, Cappello V, Vezzoli M, Rovere-Querini P, Moggio M, Ripolone M, Francolini M, Sandri M, Clementi E. Autophagy as a new therapeutic target in Duchenne muscular dystrophy. Cell Death Dis. 2012 Nov 15;3(11):e418. PMID: 23152054; PMCID: PMC3542595. DOI: https://doi.org/10.1038/cddis.2012.159
Sansa A, Hidalgo I, Miralles MP, de la Fuente S, Perez-Garcia MJ, Munell F, Soler RM, Garcera A. Spinal Muscular Atrophy autophagy profile is tissue-dependent: differential regulation between muscle and motoneurons. Acta Neuropathol Commun. 2021 Jul 3;9(1):122. PMID: 34217376; PMCID: PMC8254901. DOI: https://doi.org/10.1186/s40478-021-01223-5
Boyle KB, Randow F. The role of 'eat-me' signals and autophagy cargo receptors in innate immunity. Curr Opin Microbiol. 2013 Jun;16(3):339-48. Epub 2013 Apr 23. PMID: 23623150. DOI: https://doi.org/10.1016/j.mib.2013.03.010
Wang C, Zhao B, Zhai J, Wang A, Cao N, Liao T, Su R, He L, Li Y, Pei X, Jia Y, Yue W. Clinical-grade human umbilical cord-derived mesenchymal stem cells improved skeletal muscle dysfunction in age-associated sarcopenia mice. Cell Death Dis. 2023 May 12;14(5):321. PMID: 37173309; PMCID: PMC10182022. DOI: https://doi.org/10.1038/s41419-023-05843-8
How to Cite

This work is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License.
PAGEPress has chosen to apply the Creative Commons Attribution NonCommercial 4.0 International License (CC BY-NC 4.0) to all manuscripts to be published.