Autophagy increase in Merosin-Deficient Congenital Muscular Dystrophy type 1A
Accepted: 15 June 2023
HTML: 9
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Authors
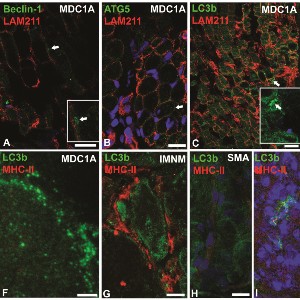
The autophagy process recycles dysfunctional cellular components and protein aggregates by sequestering them in autophagosomes directed to lysosomes for enzymatic degradation. A basal level of autophagy is essential for skeletal muscle maintenance. Increased autophagy occurs in several forms of muscular dystrophy and in the merosin-deficient congenital muscular dystrophy 1A mouse model (dy3k/dy3k) lacking the laminin-α2 chain. This pilot study aimed to compare autophagy marker expression and autophagosomes presence using light and electron microscopes and western blotting in diagnostic muscle biopsies from newborns affected by different congenital muscular myopathies and dystrophies. Morphological examination showed dystrophic muscle features, predominance of type 2A myofibers, accumulation of autophagosomes in the subsarcolemmal areas, increased number of autophagosomes overexpressing LC3b, Beclin-1 and ATG5, in the merosin-deficient newborn suggesting an increased autophagy. In Duchenne muscular dystrophy, nemaline myopathy, and spinal muscular atrophy the predominant accumulation of p62+ puncta rather suggests an autophagy impairment.




How to Cite

This work is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License.
PAGEPress has chosen to apply the Creative Commons Attribution NonCommercial 4.0 International License (CC BY-NC 4.0) to all manuscripts to be published.